
A Case of Whole Genome Analysis of SARS-CoV-2 Using Oxford Nanopore MinION System
Department of Laboratory Medicine, Hallym University College of Medicine, Seoul, Korea
ABSTRACT
The application of whole genome sequencing on SARS-CoV-2 viral genome is essential for our understanding of the molecular epidemiology and spread of viruses in the community. The portable whole genome sequencer MinION (Oxford Nanopore Technologies, ONT, UK) could be feasibly used in a clinical microbiology laboratory without the need of vast resources or stringent operating conditions. We used the MinION sequencer to analyze the viral genome sequence of one SARS-CoV-2 strain. In June 2020, nasopharyngeal specimen from one patient was subjected to whole-genome analysis using the nCoV-2019 sequencing protocol v2 of ARTIC using the MinION sequencer. The ONT MinKNOW software, RAMPART tool, and Genome Workbench were used. We identified 11 nucleotide variants using the Wuhan-Hu-1 isolate (NC_045512.2) as the reference sequence. There were six nucleotide variants (T265I, F924, Y3884L, P4715L, L5462, and Q6804L) in the ORF1ab region, one variant (D614G) in the S gene, one variant (Q57H) in ORF3a, one variant (P302) in the N gene, and two variants in each the 5′-UTR and 3′-UTR. In this prolonged coronavirus disease 2019 (COVID-19) pandemic season, the MinION system that operates an amplicon-based whole-genome sequencing protocol could be a rapid and reliable sequencer without the need of cumbersome viral cultivation.
Keywords
Nanopore sequencing, SARS-CoV-2, COVID-19, Whole genome sequencing
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)는 2019년 12월 중국 우한시에서 처음 보고되었으며 약 30,000개의 염기서열을 가진 RNA virus로서, 16개의 비구조 단백질을 만드는 open reading frames (ORFs) 1-10과 구조 단백질을 만드는 S, E, M, N 등의 구조로 이루어져 있다[1].
SARS-CoV-2 유전체 분석은 역학적 분석 및 변이형 확인을 위해 중요한 역할을 하며[2], next generation sequencing (NGS) 기기인 Illumina (San Diego, CA, USA)의 다양한 기종이나 Oxford Nanopore Technologies (ONT, Oxford, UK)의 여러 기종을 이용할 수 있다[3-7].
NGS 기기 중 ONT MinION은 본체가 손바닥 안에 들어갈 정도로 작으며, 해당사의 클라우드 기반 프로그램을 이용하면 실시간으로 분석이 가능한 장비로서, NGS를 일상적으로 수행하기 어려운 검사실 환경에서도 사용이 가능하다. 또한, 영국의 ARTIC network에서는 다양한 bioinformatic 도구를 제공하며, 아프리카의 Ebola virus 유전체 분석에 사용할 수 있는 체계가 있으며, Lassa virus, Yellow fever virus, In.uenza virus, SARS-CoV-2 등의 유전체 분석에도 사용하고 있다 [1,8]. Real-time reverse transcription (RT)- polymerase chain reaction (PCR) 시 측정된 cycle threshold (Ct) 값에 따라 RNA 양을 조절하는데, Ct 값 18-35의 검체는 농도를 희석없이 사용한다고 제시하고 있다. ARTIC network의 분석체계는 cDNA 합성 후 218개 PCR primer로 98개 유전자 부위를 증폭하는 multiplex PCR을 2개의 PCR tube로 시행한 다음, library를 제작한 뒤에 MinION 장비로 분석한다. 노트북이나 개인용 컴퓨터로 ONT에서 제공하는 MinKNOW 소프트웨어를 가동하여 염기서열 분석을 진행하며 FAST5 파일을 생성한다[8]. ARTIC network (https://artic.network/ncov-2019)에서 제공하는 ARTIC MinION 체계를 이용하여 BAM 파일, consensus.fasta 등의 파일을 생성하여 유전체 정보를 얻는다. Guppy 소프트웨어를 통해 basecalling, demultiplexing, read filtering를 할 수 있으며, RAMPART를 사용하면 실시간으로 각 유전자 부위의 read 생성을 실시간으로 확인할 수 있다.
이 연구는 2020년 6월 SARS-CoV-2 양성인 한 검체에서 ONT사의 Nanopore NGS 기기와 ARTIC MinION 체계를 사용하여 SARS-CoV-2 유전체를 분석하였다.
CASE REPORT
2020년 6월 한 대학병원에서 SARS-CoV-2 양성을 보인 비인두도말 한 검체를 사용하여, 2020년 7월 MinION 분석을 시행하였다. 환자는 국내거주자였으며, Allplex 2019-nCovV Assay (Seegene, Seoul, Korea)에서 양성으로 판정하였으며, Ct 값은 E 유전자 22.1, RdRP 유전자 24.2, N 유전자 26.4를 보였다.
사용한 검사법은 2020년 4월 9일 발표된 nCoV-2019 sequencing protocol v2 (GunIt) V.2을 위주로 하였으며, V3 primer set을 사용하였다(https://www.protocols.io/view/ncov-2019-sequencing-protocol-v2-bdp7i5rn?version_warning=no). 당시에 검체 수가 적어, 동일한 검체 1개를 5개의 barcode로 반복하여 염기서열 분석을 진행하였다. 간략하게 기술하면, RNA 11 μL와 random hexamer (50 μM) 1 μL, dNTP (10 mM) 1 μL로 65도, 5분간 반응시키고, SSIV buffer 4 μL, DTT (100 mM) 1 μL, RNase Inhibitor 1 μL, reverse transcriptase 1 μL를 혼합하여 cDNA를 합성하였다. PCR 반응은 Nanopore protocol (ver. PTC_9096_v109_revE06Feb2020)에 따라 기존 시약으로 시행하였으며, PCR tube 1, 2에 cDNA 2.5 μL, Hot Start High-Fidelity 2X master mix 12.5 μL를 분주한 뒤, V3 primer set 1 (110개 primer), set 2 (108개 primer)를 각각 3.7 μL씩 분주한 뒤 distilled water (DW)를 가하여 25 μL로 맞춘 다음 multiplex PCR을 시행하였다.
PCR 반응물을 end preparation, barcode ligation, bead purification 등으로 정제한 후 DNA 양을 측정하고 adaptor ligation 시키고, bead purification 등으로 정제한 뒤에 DNA 양을 맞추어 MinION .ow cell에 주입하였다.
MinKNOW 프로그램을 시행하여 NGS 실험을 진행하였다. ARTIC network에서 제공하는 RAMPART 프로그램을 시행하여 실시간으로 분석되는 각 유전자 부위의 read 수를 확인하였으며, reference sequence로 SARS-CoV-2 isolate Wuhan-Hu-1 (NC_045512.2)를 사용하였다.
MinKNOW에서 얻어진 FAST5 파일을 Guppy 등을 사용하여 basecalling, read filtering을 시행한 뒤, ARTIC network에서 제공하는 artic-ncov2019 프로그램으로 분석하여 BAM 파일을 구하였다. 표준염기서열로 유전자의 variant 확인을 위해 The National Center for Biotechnology Information (NCBI) Genome Workbench (version 3.6.0)를 사용하여 육안으로 분석하였다.
하나의 검체를 시험적으로 5개의 barcode로 각각 진행하였고, RAMPART 로 실시간으로 관측하면서 검체당 약 300,000개 read를 목표로 읽었으며, 표준염기서열(NC_045512.2)에 mapping된 read 수는 277,250-414,704개였다. Artic-ncov2019로 생성된 BAM 파일을 NCBI Genome Workbench로 분석하였고, variant를 육안으로 판독할 수 있었다(Fig. 1). 염기서열 차이는 표준염기서열에 대해 11개가 관찰되었으며(Table 1), ORF1ab 부위에서 T265I, F924, Y3884L, P4715L, L5462, Q6804L 등 6개 변이, S 유전자에서 D614G 변이, ORF3a 부위에서 Q57H 변이, N 유전자에서 P302 가 관찰되었으며, 5′-UTR과 3′-UTR에서 각각 1개의 유전자 염기서열 차이가 관찰되었다.
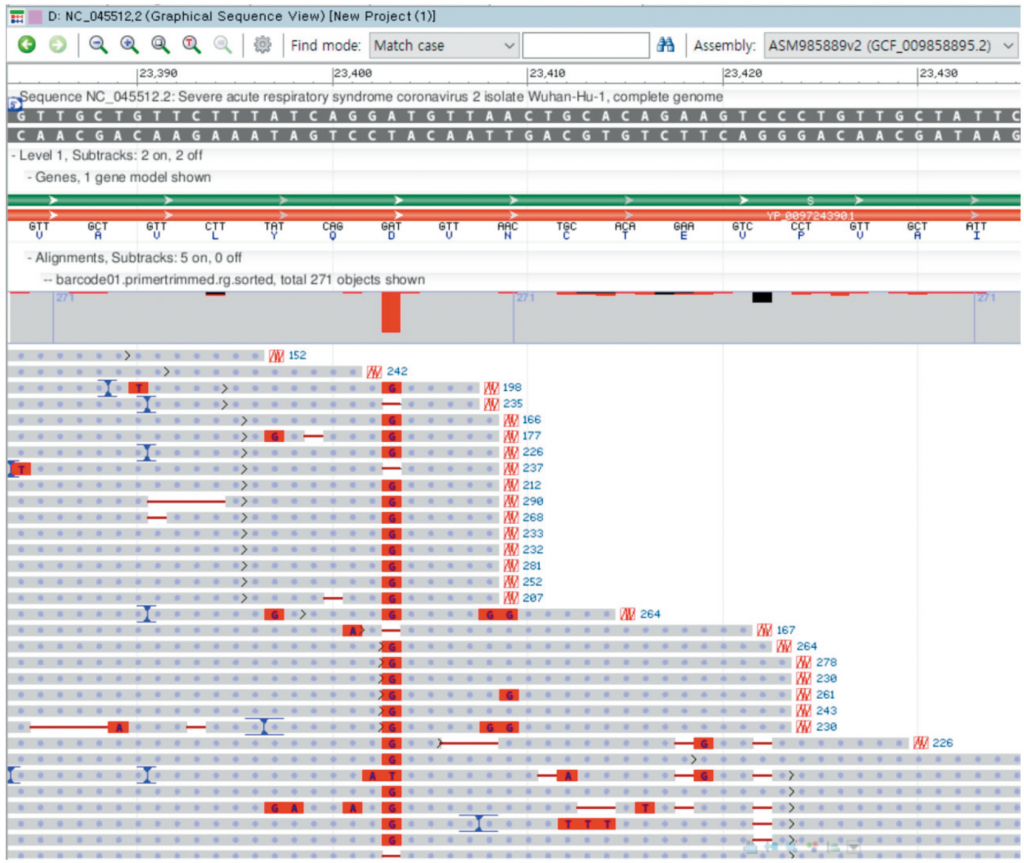
Fig. 1. Graphic sequence view of NCBI Genome Workbench for D614G variant in S gene. MinION sequencer is known as a next-generation sequencer with less accuracy, but it revealed sufficient accuracy for the mutation with amplicon-based approach. NCBI, The National Center for Biotechnology Information
Table 1. Genomic and amino acid changes of the SARS-CoV-2 positive specimen
|
Genomic change |
Amino acid change |
Gene/region |
|
241 C -> T |
|
5′-UTR |
|
1059 C -> T |
T265I |
orf1ab |
|
3037 C -> T |
F924 |
orf1ab |
|
11916 C -> T |
Y3884L |
orf1ab |
|
14408 C -> T |
P4715L |
orf1ab |
|
16650 C -> T |
L5462 |
orf1ab |
|
20675 A -> T |
Q6804L |
orf1ab |
|
23403 A -> G |
D614G |
S |
|
25563 G -> T |
Q57H |
orf3a |
|
29179 G -> T |
P302 |
N |
|
29779 G -> T |
|
3′-UTR |
신종바이러스 질환 발생시 바이러스 유전체 분석은 역학조사와 변이형 발생, 분자진단검사법의 정확도 확인 등에서 중요한 역할을 한다. 분자진단검사법에서 사용하는 primer나 probe가 부착되는 부위에 변이가 발생하거나, 백신, 항체치료제의 작용부위에 변이가 발생하는 경우를 대비하여 유전자 염기서열분석이 필요하며, whole genome sequencing (WGS) 분석이 유용하다[1,2,9]. 신종질환인 SARS-CoV-2의 경우에도 초기에 바이러스의 특성을 알기 어려웠으나, 여러 주요 변이형에 따라서 전염력, 예후, 백신의 효과 등이 달라질 수 있으므로, 이러한 변이형에 대한 이해가 필요하다[2].
MinION NGS는 long-read sequencing 기기로서, Illumina 사의 short read sequencing 기기에 비해 정확도가 떨어진다고 알려져 있으나, amplification 후 충분한 수의 read를 판독하게 되면 정확도에 문제가 없는 것으로 알려져 있다[7]. 이번 연구 대상은 본 병원에서 확인된 2번째 환자로서 검체가 많지 않은 시점이라서, 동일 검체에 다른 barcode를 붙여서 염기서열을 반복적으로 분석하였고, 육안으로 각각의 BAM 파일을 확인하였다.
MinION NGS는 비교적 소량의 검체를 처리할 수 있어서, 다른 NGS 기기에 비해 장점이 있으나, PCR amplification을 primer 200개 이상으로 시행해야 하고 NGS 시약이 소요되므로 분석에는 적지 않은 비용이 소요된다. 또한 NGS 검사에 필요한 bioinformatics 소프트웨어는 리눅스 환경이나, Guppy, RAMBART 등의 사용법에 대해 숙지가 필요하므로, 마이크로소프트 윈도우 같은 Graphic User Interface 환경에 익숙한 경우 초기에 어려움을 겪을 수 있다. 하지만, ARTIC network에 실험 및 분석 과정을 비교적 자세하게 기술하고 있어 기본적인 사용법을 익히면 적용 가능하다. Variant를 판독하는 과정은 ARTIC에서 권하는 Tablet의 경우 다루기 어려운 점이 있어 NCBI Genome Workbench를 사용하여 어렵지 않게 variant 판정이 가능하였다(Fig. 1). MinION은 소량의 검체로도 분석가능한 장점이 있지만, 필요시 대량의 검체도 처리할 수 있다. 이번 연구는 2020년 7월에 분석을 시행하여 nCoV-2019 sequencing protocol v2 (GunIt) V.2을 사용하였으나, 2020년 8월 nCoV-2019 sequencing protocol v3 (LoCost) 로 일부 시약과 절차가 변경되어 11-95개 검체를 시행할 수 있는 대량 검사 체계로 보완되었다.
이 연구의 검체는 2020년 6월의 SARS-CoV-2 real-time RT-PCR에 양성을 보인 것으로, 주요 변이로 S 유전자의 D614G와, N 유전자의 P302가 검출되었다. 이 균주는 우리나라에서만 주로 검출되는 PANGO lineage B.1.497 (이전에는 B.1.3.1로 알려짐)와 동일한 균주이며, 이 lineage는 국내에서 2020년 5월경 처음 발견된 이후, 발생이 증가하여 2021년 1월에는 전체 균주의 80%를 차지하고 있다[4, 6]. 이 균주는 2020년 2월 대구에서의 집단 발병에[10] 맞물려서 국내에서 2020년 2-3월에 유행한 PANGO lineage B.41과는 다른 종류이다.
N 유전자부위에 있는 G29179T 변이는 아미노산 변화는 없지만, CDC (Centers for Disease Control and Prevention)의 PCR primer 중 2019-nCoV-N2-F 부위에 발생하여 일부 분자진단시약의 N 유전자 검출 성능에 영향을 줄 수 있다[11] (https://primer-monitor.neb.com/ ; accessed 2021.6.23). 이 변이는 주로 우리나라에서 보고되고 있지만, 필리핀, 이스라엘, 덴마크, 캐나다, 영국에서도 빈도가 적지만 보고가 있다.
이 연구에서는 SARS-CoV-2 양성 검체로 MinION을 이용하여 유전체 분석을 진행하였으며, 우리나라에서 주로 분리되는 B.1.497 lineage로 판정할 수 있었다. NGS 기기 사용이 어려운 곳이나 소량의 검체를 분석할 경우 MinION NGS 기기는 SARS-CoV-2 유전체 분석에 유용할 것으로 생각한다.
요약
SARS-CoV-2의 유전체 분석은 분자역학을 이해하거나 지역사회의 바이러스 전염을 이해하는데 필요하다. 유전자 염기서열분석 기기인 MinION (Oxford Nanopore Technologies, ONT, UK)를 이용하면 다양한 환경에서도 검사를 할 수 있으며, 시설과 자원이 제한된 임상미생물검사실에서도 시행할 수 있다. 이 연구에서는 국내의 한 환자에서 검출된 SARS-CoV-2의 유전체 분석을 MinION 유전자염기서열 분석기로 분석하였다. 2020년 6월 서울의 한 대학병원에서 SARS-CoV-2 양성을 보인 비인두도말 검체 하나로 MinION 기기와 ARTIC nCoV-2019 sequencing protocol v2를 이용하여 유전체 염기서열 분석을 시행하였다. ONT MinKNOW, RAMPART, Genome Workbench 등의 소프트웨어를 사용하여 변이형을 확인하였다. SARS-CoV-2 표준염기서열인 Wuhan-Hu-1 isolate (NC_045512.2)에 대해 분석한 결과 11개의 유전자 변이가 관찰되었으며, ORF1ab 부위에서 6개(T265I, F924, Y3884L, P4715L, L5462, Q6804L), S 유전자에서 1개(D614G), ORF3a 부위에서 1개(Q57H), N 유전자에서 1개(P302), 5′-UTR 와 3′-UTR에서 각각 1개씩 관찰되었다. 유전자 증폭을 통한 유전체 분석이 가능한 MinION 기기는 바이러스 유행시 배양시설이 없는 곳에서 유용한 기법으로서 유전체 분석에 사용할 수 있을 것으로 보인다.
CONFLICTS OF INTEREST
No potential con.icts of interest relevant to this article were reported.
FUNDING
이 연구는 한림의대 산학협력단의 연구비 지원과 보건복지부의 재원으로 한국보건산업진흥원의 보건의료기술 연구개발사업 지원에 의하여 이루어졌습니다(Grant No. HI20C0071).
REFERENCES
1. Uhteg K, Carroll KC, Mostafa HH. Coronavirus detection in the clinical microbiology laboratory: are we ready for identifying and diagnosing a novel virus? Clin Lab Med 2020;40:459-72. 


2. Burki T. Understanding variants of SARS-CoV-2. Lancet 2021;397:462.
3. Kim HM, Jeon S, Chung O, Jun JH, Kim HS, Blazyte A, et al. Comparative analysis of 7 short-read sequencing platforms using the Korean Reference Genome: MGI and Illumina sequencing benchmark for whole-genome sequencing. Gigascience 2021;10: giab014.
4. Kim JM, Park SY, Lee D, Kim JS, Park Y, Gwack J, et al.Genomic investigation of the coronavirus disease-2019 outbreak in the Republic of Korea. Sci Rep 2021;11:1-10.
5. Hourdel V, Kwasiborski A, Baliere C, Matheus S, Batejat CF, Manuguerra JC, et al.Rapid genomic characterization of SARS-CoV-2 by direct amplicon-based sequencing through comparison of MinION and Illumina iSeq100(TM) system. Front Microbiol 2020;11:571328.
6. Park AK, Kim IH, Kim J, Kim JM, Kim HM, Lee CY, et al.Genomic surveillance of SARS-CoV-2: distribution of clades in the Republic of Korea in 2020. Osong Public Health Res Perspect 2021;12:37-43.
7. Bull RA, Adikari TN, Ferguson JM, Hammond JM, Stevanovski I, Beukers AG, et al.Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat Commun 2020;11:6272.
8. Tyson JR, James P, Stoddart D, Sparks N, Wickenhagen A, Hall G, et al.Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore.
9. Tali SHS, LeBlanc JJ, Sadiq Z, Oyewunmi OD, Camargo C, Nikpour B, et al.Tools and techniques for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)/COVID-19 detection. Clin Microbiol Rev 2021;34:e00228-20.
10. Park S, Kim DH, Lee WM, Ha JS, Jeon DS, Lee JH, et al.Experience at department of laboratory medicine during the COVID-19 outbreak in Daegu. Ann Clin Microb 2020;23:225-31.
11. Hong KH, In JW, Lee J, Kim SY, Lee KA, Kim S, et al.Prevalence of a single-nucleotide variant of SARS-CoV-2 in Korea and its impact on the diagnostic sensitivity of the Xpert Xpress SARS-CoV-2 assay. Ann Lab Med 2022;42:96-9.