
Minh Phuong Trinh![]() , Min-Kyoung Shin
, Min-Kyoung Shin![]()

1. Turenne CY. Nontuberculous mycobacteria: insights on taxonomy and evolution. Infect Genet Evol 2019;72:159-68.

2. Forbes BA, Hall GS, Miller MB, Novak SM, Rowlinson MC, Salfinger M, et al. Practical guidance for clinical microbiology laboratories: mycobacteria. Clin Microbiol Rev 2018;31:e00038-17. 
3. WHO. Global tuberculosis report 2023. Geneva: World Health Organization; 2023.
4. Zhang H, Tang M, Li D, Xu M, Ao Y, Lin L. Applications and advances in molecular diagnostics: revolutionizing non-tuberculous mycobacteria species and subspecies identification. Front Public Health 2024;12:1410672.
5. Griffith DE and Aksamit TR. Understanding nontuberculous mycobacterial lung disease: it’s been a long time coming. F1000Res 2016;5:2797.
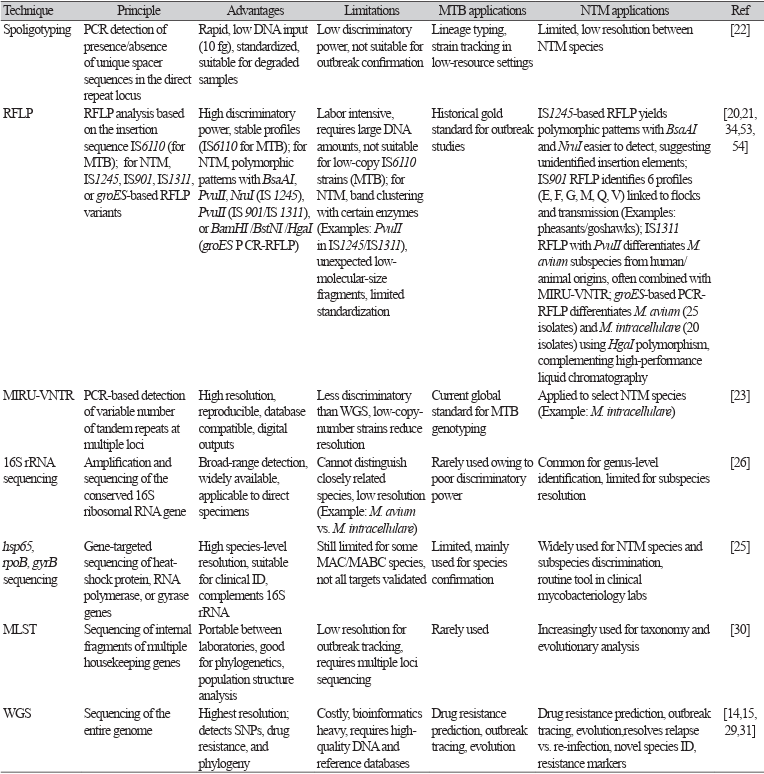
6. Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rüsch-Gerdes S, Willery E, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unitvariable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol 2006;44:4498-510.
7. Couvin D, Allaguy AS, Ez-Zari A, Jagielski T, Rastogi N. Molecular typing of Mycobacterium tuberculosis: a review of current methods, databases, softwares, and analytical tools. FEMS Microbiol Rev 2025;49:fuaf017.
8. Baldwin SL, Larsen SE, Ordway D, Cassell G, Coler RN. The complexities and challenges of preventing and treating nontuberculous mycobacterial diseases. PLoS Negl Trop Dis 2019;13:e0007083.
9. WHO. Global tuberculosis report 2024. Geneva: World Health Organization; 2024. p. 68.
10. Churchyard G, Kim P, Shah NS, Rustomjee R, Gandhi N, Mathema B, et al. What we know about tuberculosis transmission: an overview. J Infect Dis 2017;216:S629-35.
11. Auld SC, Kasmar AG, Dowdy DW, Mathema B, Gandhi NR, Churchyard GJ, et al. Research roadmap for tuberculosis transmission science: where do we go from here and how will we know when we’re there? J Infect Dis 2017;216:S662-8.
12. Nahid P, Mase SR, Migliori GB, Sotgiu G, Bothamley GH, Brozek JL, et al. Treatment of drug-resistant tuberculosis an official ATS/CDC/ERS/IDSA clinical practice guideline. Am J Respir Crit Care Med 2019;200:E93-142.
13. Dheda K, Cox H, Esmail A, Wasserman S, Chang KC, Lange C. Recent controversies about MDR and XDR-TB: global implementation of the WHO shorter MDR-TB regimen and bedaquiline for all with MDR-TB? Respirology 2018;23:36-45.
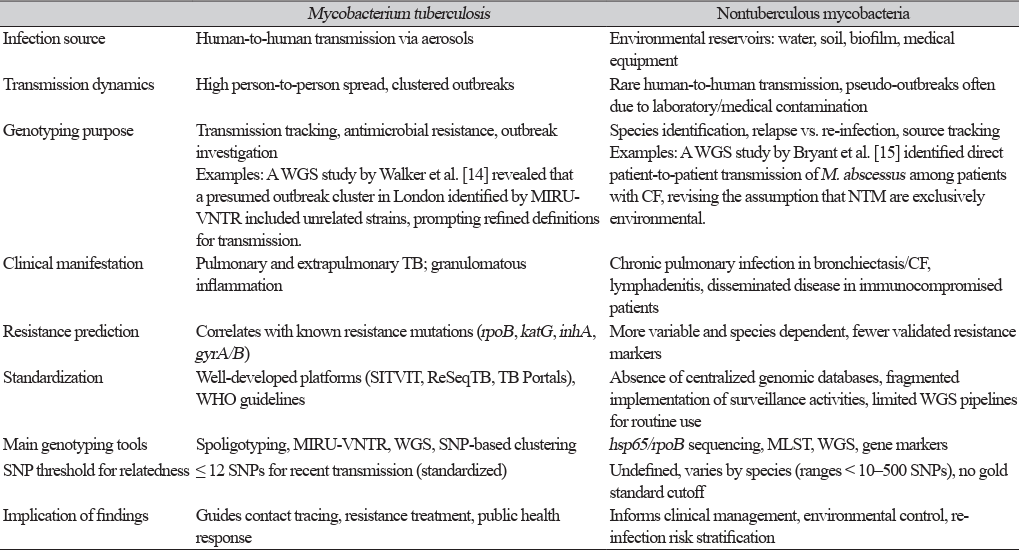
14. Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis 2013;13:137-46.
15. Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 2013;381:1551-60.
16. Fox GJ, Barry SE, Britton WJ, Marks GB. Contact investigation for tuberculosis: a systematic review and meta-analysis. Eur Respir J 2013;41:140-56.
17. Kremer K, van Soolingen D, Frothingham R, Haas WH, Hermans PW, Martín C, et al. Comparison of methods based on different molecular epidemiological markers for typing of Mycobacterium tuberculosis complex strains: interlaboratory study of discriminatory power and reproducibility. J Clin Microbiol 1999;37:2607-18.
18. Braden CR, Crawford JT, Schable BA. Quality assessment of Mycobacterium tuberculosis genotyping in a large laboratory network. Emerg Infect Dis 2002;8:1210-5.
19. de Boer AS, Borgdorff MW, de Haas PE, Nagelkerke NJ, van Embden JD, van Soolingen D. Analysis of rate of change of IS6110 RFLP patterns of Mycobacterium tuberculosis based on serial patient isolates. J Infect Dis 1999;180:1238-44.
20. Garzelli C, Lari N, Nguon B, Cavallini M, Pistello M, Falcone G. Comparison of three restriction endonucleases in IS1245-based RFLP typing of Mycobacterium avium. J Med Microbiol 1997;46:933-9.
21. Aravindhan V, Sulochana S, Narayanan S, Paramasivam CN, Narayanan PR. Identification & differentiation of Mycobacterium avium & M. intracellulare by PCR- RFLP assay using the groES gene. Indian J Med Res 2007;126:575-9.
22. Molina-Moya B, Gomgnimbou MK, Spinasse L, Obasanya J, Oladimeji O, Dacombe R, et al. Mycobacterium tuberculosis complex genotypes circulating in Nigeria based on spoligotyping obtained from Ziehl-Neelsen stained slides extracted DNA. PLoS Negl Trop Dis 2018;12:e0006242.
23. Ichikawa K, Yagi T, Inagaki T, Moriyama M, Nakagawa T, Uchiya KI, et al. Molecular typing of Mycobacterium intracellulare using multilocus variable-number of tandem-repeat analysis: identification of loci and analysis of clinical isolates. Microbiology 2010;156(Pt 2):496-504.
24. Adékambi T, Colson P, Drancourt M. rpoB-based identification of nonpigmented and latepigmenting rapidly growing mycobacteria. J Clin Microbiol 2003;41:5699-708.
25. Kim SH and Shin JH. Identification of nontuberculous mycobacteria using multilocous sequence analysis of 16S rRNA, hsp65, and rpoB. J Clin Lab Anal 2018;32:e22184.
26. Vos M, Quince C, Pijl AS, de Hollander M, Kowalchuk GA. A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS One 2012;7:e30600.
27. Comas I and Gagneux S. The past and future of tuberculosis research. PLoS Pathog 2009;5:e1000600.
28. WHO. Molecular line probe assays for rapid screening of patients at risk of multidrug-resistant tuberculosis (MDR-TB). Geneva: World Health Organization; 2008.
29. Nikolayevskyy V, Niemann S, Anthony R, van Soolingen D, Tagliani E, Ködmön C, et al. Role and value of whole genome sequencing in studying tuberculosis transmission. Clin Microbiol Infect 2019;25:1377-82.
30. Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol 2013;11:72836.
31. Stucki D and Gagneux S. Single nucleotide polymorphisms in Mycobacterium tuberculosis and the need for a curated database. Tuberculosis 2013;93:30-9.
32. Roetzer A, Diel R, Kohl TA, Rückert C, Nübel U, Blom J, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med 2013;10:e1001387.
33. Coll F, McNerney R, Preston MD, Guerra-Assunção JA, Warry A, Hill-Cawthorne G, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med 2015;7:51.
34. Tortoli E, Meehan CJ, Grottola A, Fregni Serpini G, Fabio A, Trovato A, et al. Genome-based taxonomic revision detects a number of synonymous taxa in the genus Mycobacterium. Infect Genet Evol 2019;75:103983.
35. Phelan JE, O’Sullivan DM, Machado D, Ramos J, Oppong YEA, Campino S, et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med 2019;11:41.
36. Gagneux S. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol 2018;16:202-13.
37. Lipworth S, Hough N, Leach L, Morgan M, Jeffery K, Andersson M, et al. Whole-genome sequencing for predicting clarithromycin resistance in Mycobacterium abscessus. Antimicrob Agents Chemother 2018;63:e01204-18.
38. Brown-Elliott BA, Nash KA, Wallace RJ Jr. Antimicrobial susceptibility testing, drug resistance mechanisms, and therapy of infections with nontuberculous mycobacteria. Clin Microbiol Rev 2012;25:545-82.
39. Diel R, Lipman M, Hoefsloot W. High mortality in patients with Mycobacterium avium complex lung disease: a systematic review. BMC Infect Dis 2018;18:206.
40. Doyle RM, Burgess C, Williams R, Gorton R, Booth H, Brown J, et al. Direct whole-genome sequencing of sputum accurately identifies drug-resistant Mycobacterium tuberculosis faster than MGIT culture sequencing. J Clin Microbiol 2018;56:e00666-18.
41. Rosenthal A, Gabrielian A, Engle E, Hurt DE, Alexandru S, Crudu V, et al. The TB portals: an open-access, web-based platform for global drug-resistant-tuberculosis data sharing and analysis. J Clin Microbiol 2017;55:3267-82.
42. Tortoli E. Microbiological features and clinical relevance of new species of the genus Mycobacterium. Clin Microbiol Rev 2014;27:727-52.
43. van Ingen J, Kohl TA, Kranzer K, Hasse B, Keller PM, Katarzyna Szafrańska A, et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect Dis 2017;17:1033-41.
44. Deelder W, Christakoudi S, Phelan J, Benavente ED, Campino S, McNerney R, et al. Machine learning predicts accurately Mycobacterium tuberculosis drug resistance from whole genome sequencing data. Front Genet 2019;10:922.
45. Yang Y, Niehaus KE, Walker TM, Iqbal Z, Walker AS, Wilson DJ, et al. Machine learning for classifying tuberculosis drug-resistance from DNA sequencing data. Bioinformatics 2018;34:1666-71.
46. Meehan CJ, Goig GA, Kohl TA, Verboven L, Dippenaar A, Ezewudo M, et al. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat Rev Microbiol 2019;17:533-45.
47. Votintseva AA, Bradley P, Pankhurst L, Del Ojo Elias C, Loose M, Nilgiriwala K, et al. Same-day diagnostic and surveillance data for tuberculosis via whole-genome sequencing of direct respiratory samples. J Clin Microbiol 2017;55:1285-98.
48. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007;315:1709-12.
49. Chakravorty S, Simmons AM, Rowneki M, Parmar H, Cao Y, Ryan J, et al. The new Xpert MTB/RIF ultra: improving detection of Mycobacterium tuberculosis and resistance to rifampin in an assay suitable for point-of-care testing. mBio 2017;8:e00812-17.
50. Kapopoulou A, Lew JM, Cole ST. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 2011;91:8-13.
51. Chernyaeva EN, Shulgina MV, Rotkevich MS, Dobrynin PV, Simonov SA, Shitikov EA, et al. Genome-wide Mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genomics 2014;15:308.
52. CRyPTIC consortium and the 100,000 genomes project; Allix-Béguec C, Arandjelovic I, Bi L, Beckert P, Bonnet M, Bradley P, et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med 2018;379:1403-15.
53. Radomski N, Thibault VC, Karoui C, de Cruz K, Cochard T, Gutiérrez C, et al. Determination of genotypic diversity of Mycobacterium avium subspecies from human and animal origins by mycobacterial interspersed repetitive-unit-variable-number tandem-repeat and IS1311 restriction fragment length polymorphism typing methods. J Clin Microbiol 2010;48:1026-34.
54. Moravkova M, Lamka J, Slany M, Pavlik I. Genetic IS901 RFLP diversity among Mycobacterium avium subsp. avium isolates from four pheasant flocks. J Vet Sci 2013;14:99-102.
1. Turenne CY. Nontuberculous mycobacteria: insights on taxonomy and evolution. Infect Genet Evol 2019;72:159-68.
2. Forbes BA, Hall GS, Miller MB, Novak SM, Rowlinson MC, Salfinger M, et al. Practical guidance for clinical microbiology laboratories: mycobacteria. Clin Microbiol Rev 2018;31:e00038-17.
3. WHO. Global tuberculosis report 2023. Geneva: World Health Organization; 2023.
4. Zhang H, Tang M, Li D, Xu M, Ao Y, Lin L. Applications and advances in molecular diagnostics: revolutionizing non-tuberculous mycobacteria species and subspecies identification. Front Public Health 2024;12:1410672.
5. Griffith DE and Aksamit TR. Understanding nontuberculous mycobacterial lung disease: it’s been a long time coming. F1000Res 2016;5:2797.
6. Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rüsch-Gerdes S, Willery E, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unitvariable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol 2006;44:4498-510.
7. Couvin D, Allaguy AS, Ez-Zari A, Jagielski T, Rastogi N. Molecular typing of Mycobacterium tuberculosis: a review of current methods, databases, softwares, and analytical tools. FEMS Microbiol Rev 2025;49:fuaf017.
8. Baldwin SL, Larsen SE, Ordway D, Cassell G, Coler RN. The complexities and challenges of preventing and treating nontuberculous mycobacterial diseases. PLoS Negl Trop Dis 2019;13:e0007083.
9. WHO. Global tuberculosis report 2024. Geneva: World Health Organization; 2024. p. 68.
10. Churchyard G, Kim P, Shah NS, Rustomjee R, Gandhi N, Mathema B, et al. What we know about tuberculosis transmission: an overview. J Infect Dis 2017;216:S629-35.
11. Auld SC, Kasmar AG, Dowdy DW, Mathema B, Gandhi NR, Churchyard GJ, et al. Research roadmap for tuberculosis transmission science: where do we go from here and how will we know when we’re there? J Infect Dis 2017;216:S662-8.
12. Nahid P, Mase SR, Migliori GB, Sotgiu G, Bothamley GH, Brozek JL, et al. Treatment of drug-resistant tuberculosis an official ATS/CDC/ERS/IDSA clinical practice guideline. Am J Respir Crit Care Med 2019;200:E93-142.
13. Dheda K, Cox H, Esmail A, Wasserman S, Chang KC, Lange C. Recent controversies about MDR and XDR-TB: global implementation of the WHO shorter MDR-TB regimen and bedaquiline for all with MDR-TB? Respirology 2018;23:36-45.
14. Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis 2013;13:137-46.
15. Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 2013;381:1551-60.
16. Fox GJ, Barry SE, Britton WJ, Marks GB. Contact investigation for tuberculosis: a systematic review and meta-analysis. Eur Respir J 2013;41:140-56.
17. Kremer K, van Soolingen D, Frothingham R, Haas WH, Hermans PW, Martín C, et al. Comparison of methods based on different molecular epidemiological markers for typing of Mycobacterium tuberculosis complex strains: interlaboratory study of discriminatory power and reproducibility. J Clin Microbiol 1999;37:2607-18.
18. Braden CR, Crawford JT, Schable BA. Quality assessment of Mycobacterium tuberculosis genotyping in a large laboratory network. Emerg Infect Dis 2002;8:1210-5.
19. de Boer AS, Borgdorff MW, de Haas PE, Nagelkerke NJ, van Embden JD, van Soolingen D. Analysis of rate of change of IS6110 RFLP patterns of Mycobacterium tuberculosis based on serial patient isolates. J Infect Dis 1999;180:1238-44.
20. Garzelli C, Lari N, Nguon B, Cavallini M, Pistello M, Falcone G. Comparison of three restriction endonucleases in IS1245-based RFLP typing of Mycobacterium avium. J Med Microbiol 1997;46:933-9.
21. Aravindhan V, Sulochana S, Narayanan S, Paramasivam CN, Narayanan PR. Identification & differentiation of Mycobacterium avium & M. intracellulare by PCR- RFLP assay using the groES gene. Indian J Med Res 2007;126:575-9.
22. Molina-Moya B, Gomgnimbou MK, Spinasse L, Obasanya J, Oladimeji O, Dacombe R, et al. Mycobacterium tuberculosis complex genotypes circulating in Nigeria based on spoligotyping obtained from Ziehl-Neelsen stained slides extracted DNA. PLoS Negl Trop Dis 2018;12:e0006242.
23. Ichikawa K, Yagi T, Inagaki T, Moriyama M, Nakagawa T, Uchiya KI, et al. Molecular typing of Mycobacterium intracellulare using multilocus variable-number of tandem-repeat analysis: identification of loci and analysis of clinical isolates. Microbiology 2010;156(Pt 2):496-504.
24. Adékambi T, Colson P, Drancourt M. rpoB-based identification of nonpigmented and latepigmenting rapidly growing mycobacteria. J Clin Microbiol 2003;41:5699-708.
25. Kim SH and Shin JH. Identification of nontuberculous mycobacteria using multilocous sequence analysis of 16S rRNA, hsp65, and rpoB. J Clin Lab Anal 2018;32:e22184.
26. Vos M, Quince C, Pijl AS, de Hollander M, Kowalchuk GA. A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS One 2012;7:e30600.
27. Comas I and Gagneux S. The past and future of tuberculosis research. PLoS Pathog 2009;5:e1000600.
28. WHO. Molecular line probe assays for rapid screening of patients at risk of multidrug-resistant tuberculosis (MDR-TB). Geneva: World Health Organization; 2008.
29. Nikolayevskyy V, Niemann S, Anthony R, van Soolingen D, Tagliani E, Ködmön C, et al. Role and value of whole genome sequencing in studying tuberculosis transmission. Clin Microbiol Infect 2019;25:1377-82.
30. Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol 2013;11:72836.
31. Stucki D and Gagneux S. Single nucleotide polymorphisms in Mycobacterium tuberculosis and the need for a curated database. Tuberculosis 2013;93:30-9.
32. Roetzer A, Diel R, Kohl TA, Rückert C, Nübel U, Blom J, et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med 2013;10:e1001387.
33. Coll F, McNerney R, Preston MD, Guerra-Assunção JA, Warry A, Hill-Cawthorne G, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med 2015;7:51.
34. Tortoli E, Meehan CJ, Grottola A, Fregni Serpini G, Fabio A, Trovato A, et al. Genome-based taxonomic revision detects a number of synonymous taxa in the genus Mycobacterium. Infect Genet Evol 2019;75:103983.
35. Phelan JE, O’Sullivan DM, Machado D, Ramos J, Oppong YEA, Campino S, et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med 2019;11:41.
36. Gagneux S. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol 2018;16:202-13.
37. Lipworth S, Hough N, Leach L, Morgan M, Jeffery K, Andersson M, et al. Whole-genome sequencing for predicting clarithromycin resistance in Mycobacterium abscessus. Antimicrob Agents Chemother 2018;63:e01204-18.
38. Brown-Elliott BA, Nash KA, Wallace RJ Jr. Antimicrobial susceptibility testing, drug resistance mechanisms, and therapy of infections with nontuberculous mycobacteria. Clin Microbiol Rev 2012;25:545-82.
39. Diel R, Lipman M, Hoefsloot W. High mortality in patients with Mycobacterium avium complex lung disease: a systematic review. BMC Infect Dis 2018;18:206.
40. Doyle RM, Burgess C, Williams R, Gorton R, Booth H, Brown J, et al. Direct whole-genome sequencing of sputum accurately identifies drug-resistant Mycobacterium tuberculosis faster than MGIT culture sequencing. J Clin Microbiol 2018;56:e00666-18.
41. Rosenthal A, Gabrielian A, Engle E, Hurt DE, Alexandru S, Crudu V, et al. The TB portals: an open-access, web-based platform for global drug-resistant-tuberculosis data sharing and analysis. J Clin Microbiol 2017;55:3267-82.
42. Tortoli E. Microbiological features and clinical relevance of new species of the genus Mycobacterium. Clin Microbiol Rev 2014;27:727-52.
43. van Ingen J, Kohl TA, Kranzer K, Hasse B, Keller PM, Katarzyna Szafrańska A, et al. Global outbreak of severe Mycobacterium chimaera disease after cardiac surgery: a molecular epidemiological study. Lancet Infect Dis 2017;17:1033-41.
44. Deelder W, Christakoudi S, Phelan J, Benavente ED, Campino S, McNerney R, et al. Machine learning predicts accurately Mycobacterium tuberculosis drug resistance from whole genome sequencing data. Front Genet 2019;10:922.
45. Yang Y, Niehaus KE, Walker TM, Iqbal Z, Walker AS, Wilson DJ, et al. Machine learning for classifying tuberculosis drug-resistance from DNA sequencing data. Bioinformatics 2018;34:1666-71.
46. Meehan CJ, Goig GA, Kohl TA, Verboven L, Dippenaar A, Ezewudo M, et al. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat Rev Microbiol 2019;17:533-45.
47. Votintseva AA, Bradley P, Pankhurst L, Del Ojo Elias C, Loose M, Nilgiriwala K, et al. Same-day diagnostic and surveillance data for tuberculosis via whole-genome sequencing of direct respiratory samples. J Clin Microbiol 2017;55:1285-98.
48. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007;315:1709-12.
49. Chakravorty S, Simmons AM, Rowneki M, Parmar H, Cao Y, Ryan J, et al. The new Xpert MTB/RIF ultra: improving detection of Mycobacterium tuberculosis and resistance to rifampin in an assay suitable for point-of-care testing. mBio 2017;8:e00812-17.
50. Kapopoulou A, Lew JM, Cole ST. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 2011;91:8-13.
51. Chernyaeva EN, Shulgina MV, Rotkevich MS, Dobrynin PV, Simonov SA, Shitikov EA, et al. Genome-wide Mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genomics 2014;15:308.
52. CRyPTIC consortium and the 100,000 genomes project; Allix-Béguec C, Arandjelovic I, Bi L, Beckert P, Bonnet M, Bradley P, et al. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. N Engl J Med 2018;379:1403-15.
53. Radomski N, Thibault VC, Karoui C, de Cruz K, Cochard T, Gutiérrez C, et al. Determination of genotypic diversity of Mycobacterium avium subspecies from human and animal origins by mycobacterial interspersed repetitive-unit-variable-number tandem-repeat and IS1311 restriction fragment length polymorphism typing methods. J Clin Microbiol 2010;48:1026-34.
54. Moravkova M, Lamka J, Slany M, Pavlik I. Genetic IS901 RFLP diversity among Mycobacterium avium subsp. avium isolates from four pheasant flocks. J Vet Sci 2013;14:99-102.