
Takashi Takahashi![]()

1. Naik S, Kashyap D, Deep J, Darwish S, Cross J, Mansoor E, et al. Utilizing next-generation sequencing: advancements in the diagnosis of fungal infections. Diagnostics (Basel) 2024;14:1664.


2. Li W, Cui Q, Bai L, Fu P, Han H, Liu J, et al. Application of whole-genome sequencing in the national molecular tracing network for foodborne disease surveillance in China. Foodborne Pathog Dis 2021;18:538-46.
3. Gomes E, Araújo D, Nogueira T, Oliveira R, Silva S, Oliveira LVN, et al. Advances in whole genome sequencing for foodborne pathogens: implications for clinical infectious disease surveillance and public health. Front Cell Infect Microbiol 2025;15:1593219.
4. Wesenberg S, Mueller C, Nestmann F, Holthoff-Detto V. Effects of an animal-assisted intervention on social behaviour, emotions, and behavioural and psychological symptoms in nursing home residents with dementia. Psychogeriatrics 2019;19:219-27.
5. Thodberg K, Videbech PB, Hansen TGB, Pedersen AB, Christensen JW. Dog visits in nursing homes – increase complexity or keep it simple? A randomised controlled study. PLoS One 2021;16:e0251571.
6. Centers for Disease Control and Prevention. About One Health (updated on 27 June 2025). https://www.cdc.gov/onehealth/index.html [Online] (last visited on 12 September 2025).
7. Shropshire WC, Hanson BM, Shelburne SA. Genome-wide approaches to bacterial strain typing: a history and review of recent methodological advances. Curr Opin Infect Dis 2025;38:329-38.
8. Maeda T, Yoshida H, Abe N, Murakami K, Goto M, Takahashi T. Draft genome sequence of emm103/ST1363 Streptococcus pyogenes strain AB1, isolated from the blood of a woman with peritonitis and toxic shock syndrome. Microbiol Resour Announc 2024;13:e0102723.
9. Anslan S, Mikryukov V, Armolaitis K, Ankuda J, Lazdina D, Makovskis K, et al. Highly comparable metabarcoding results from MGI-Tech and Illumina sequencing platforms. PeerJ 2021;9:e12254.
10. Yoshida H, Katayama Y, Fukushima Y, Ohtaki H, Ohkusu K, Mizutani T, et al. Draft genome sequence of Streptococcus canis clinical strain OT1, isolated from a dog owner with invasive infection without a dog bite in Japan. Microbiol Resour Announc 2019;8:e00770-19.
11. Fukushima Y, Murata Y, Katayama Y, Tsuyuki Y, Yoshida H, Mizutani T, et al. Draft genome sequence of blood-origin Streptococcus canis strain FU149, isolated from a dog with necrotizing soft tissue infection. Microbiol Resour Announc 2020;9:e00737-20.
12. Kim JS, Sakaguchi S, Fukushima Y, Yoshida H, Nakano T, Takahashi T. Complete genome sequences of four Streptococcus canis strains isolated from dogs in South Korea. Microbiol Resour Announc 2020;9:e00818-20.
13. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34:i884-90.
14. Kolar O, Appleberry H, Wolfe AJ, Kula A, Putonti C. Complete genome sequence of vaginal swab isolate Enterococcus faecalis UMB6935B, including two complete plasmids. Microbiol Resour Announc 2025;14:e0036825.
15. Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A 2005;102:13950-5.
16. He EM, Chen CW, Guo Y, Hsu MH, Zhang L, Chen HL, et al. The genome of serotype VI Streptococcus agalactiae serotype VI and comparative analysis. Gene 2017;597:59-65.
17. EZBioCloud. About Pan-Genome Orthologous Group (POG) (updated on 15 May 2017). https://help.ezbiocloud.net/uncategorized/pan-genome-orthologous-group-pog/ [Online] (last visited on 12 September 2025).
18. Lin G, Chai J, Yuan S, Mai C, Cai L, Murphy RW, et al. VennPainter: a tool for the comparison and identification of candidate genes based on Venn diagrams. PLoS One 2016;11:e0154315.
19. Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016;44:D457-62.
20. Takahashi T, Lee S, Kim S. Genomic characteristics of Streptococcus agalactiae based on the pan-genome orthologous group analysis according to invasiveness and capsular genotype. J Infect Chemother 2021;27:814-9.
21. Kim JM, Fukushima Y, Yoshida H, Kim JS, Takahashi T. Comparative genomic features of Streptococcus canis based on pan-genome orthologous group analysis according to sequence type. Jpn J Infect Dis 2022;75:269-76.
22. Takahashi T, Kim H, Kim HS, Kim HS, Song W, Kim JS. Comparative genomic analysis of staphylococcal cassette chromosome mec type V Staphylococcus aureus strains and estimation of the emergence of SCCmec V clinical isolates in Korea. Ann Lab Med 2024;44:47-55.
23. CompArative GEne Cluster Analysis Toolbox (CAGECAT) project. Welcome to CAGECAT. https://cagecat.bioinformatics.nl [Online] (last visited on 12 September 2025).
24. van den Belt M, Gilchrist C, Booth TJ, Chooi YH, Medema MH, Alanjary M. The CompArative GEne Cluster Analysis Toolbox for rapid search and visualisation of homologous gene clusters. BMC Bioinformatics 2023;24:181.
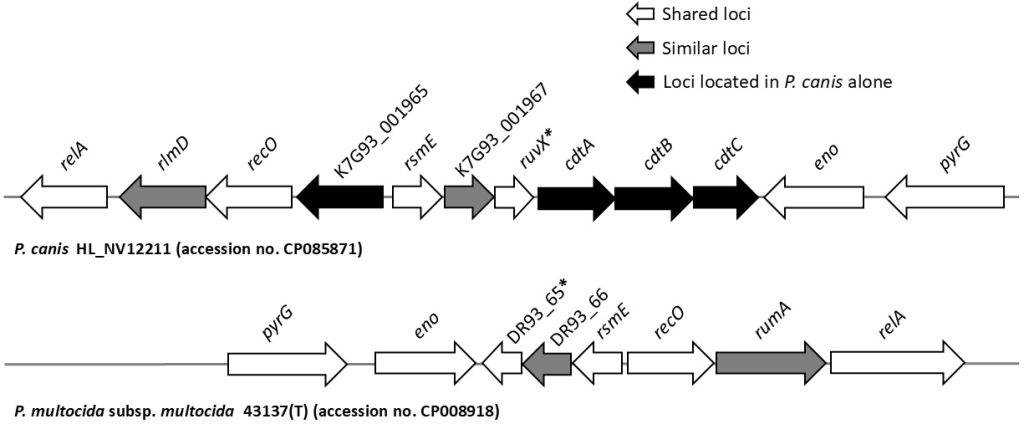
25. Yoshida H, Kim JM, Maeda T, Goto M, Tsuyuki Y, Shibata S, et al. Virulence-associated genome sequences of Pasteurella canis and unique toxin gene prevalence of P. canis and Pasteurella multocida isolated from humans and companion animals. Ann Lab Med 2023;43:263-72.
26. Virulence Factor Database (VFDB) project. Up-to-date knowledge of VFs of various bacterial pathogens. http://www.mgc.ac.cn/VFs/ [Online] (last visited on 12 September 2025).
27. Hirose Y, Yamaguchi M, Okuzaki D, Motooka D, Hamamoto H, Hanada T, et al. Streptococcus pyogenes transcriptome changes in the inflammatory environment of necrotizing fasciitis. Appl Environ Microbiol 2019;85:e01428-19.
28. Hirose Y, Yamaguchi M, Sumitomo T, Nakata M, Hanada T, Okuzaki D, et al. Streptococcus pyogenes upregulates arginine catabolism to exert its pathogenesis on the skin surface. Cell Rep 2021;34:108924.
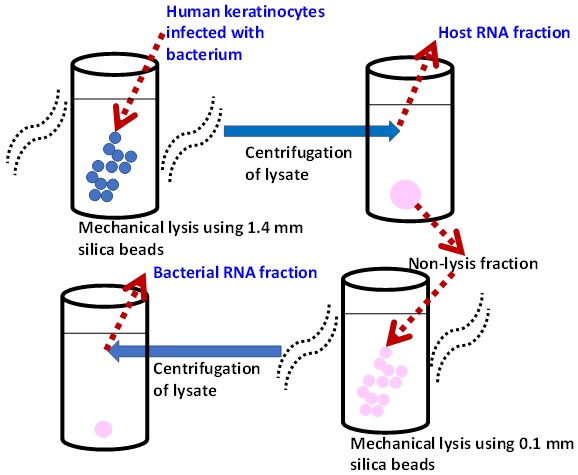
29. Yoshida H, Goto M, Tsuyuki Y, Kim JS, Takahashi T. Streptococcus canis transcriptomic modifications in host cell entry environments of human keratinocytes. BMC Genomics 2024;25:1028.
30. Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 2012;10:618-30.
31. Deb S, Basu J, Choudhary M. An overview of next generation sequencing strategies and genomics tools used for tuberculosis research. J Appl Microbiol 2024;135:lxae174.
32. Wilkening RV, Langouët-Astrié C, Severn MM, Federle MJ, Horswill AR. Identifying genetic determinants of Streptococcus pyogenes-host interactions in a murine intact skin infection model. Cell Rep 2023;42:113332.
33. Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 1998;95:3140-5.
34. Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 2010;11:595.
35. Uelze L, Grützke J, Borowiak M, Hammerl JA, Juraschek K, Deneke C, et al. Typing methods based on whole genome sequencing data. One Health Outlook 2020;2:3.
36. Silva M, Machado MP, Silva DN, Rossi M, Moran-Gilad J, Santos S, et al. chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb Genom 2018;4:e000166.
37. Seah YM, Stewart MK, Hoogestraat D, Ryder M, Cookson BT, Salipante SJ, et al. In silico evaluation of variant calling methods for bacterial whole-genome sequencing assays. J Clin Microbiol 2023;61:e0184222.
38. Hall MB, Wick RR, Judd LM, Nguyen AN, Steinig EJ, Xie O, et al. Benchmarking reveals superiority of deep learning variant callers on bacterial nanopore sequence data. eLife 2024;13:RP98300.
39. Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022;399:629-55.
40. Sherry NL, Lee JYH, Giulieri SG, Connor CH, Horan K, Lacey JA, et al. Genomics for antimicrobial resistance-progress and future directions. Antimicrob Agents Chemother 2025;69:e0108224.
41. Doyle RM, O’Sullivan DM, Aller SD, Bruchmann S, Clark T, Coello Pelegrin A, et al. Discordant bioinformatic predictions of antimicrobial resistance from whole-genome sequencing data of bacterial isolates: an inter-laboratory study. Microb Genom 2020;6:e000335.
42. Burrus V, Pavlovic G, Decaris B, Guédon G. Conjugative transposons: the tip of the iceberg. Mol Microbiol 2002;46:601-10.
43. Huang J, Liang Y, Guo D, Shang K, Ge L, Kashif J, et al. Comparative genomic analysis of the ICESa2603 family ICEs and spread of erm(B)- and tet(O)-carrying transferable 89K-subtype ICEs in swine and bovine isolates in China. Front Microbiol 2016;7:55.
44. Beres SB, Musser JM. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One 2007;2:e800.
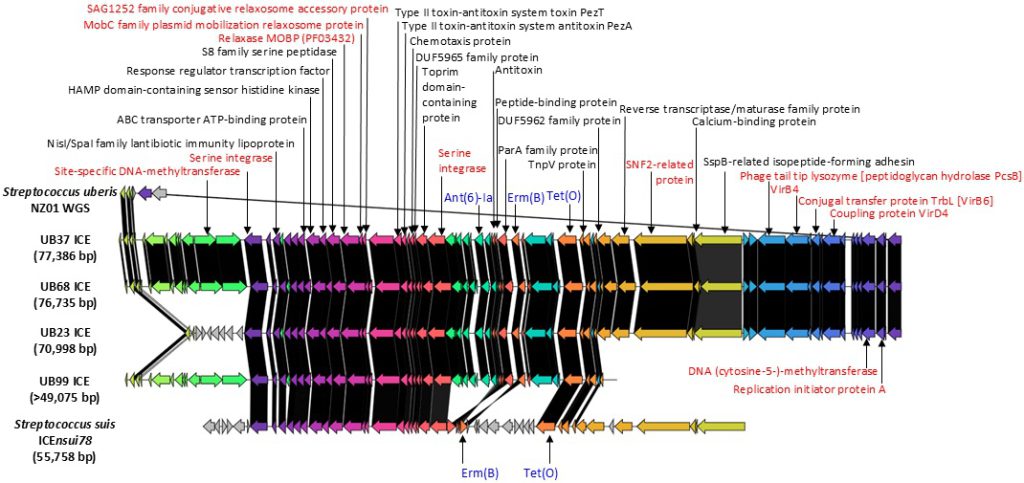
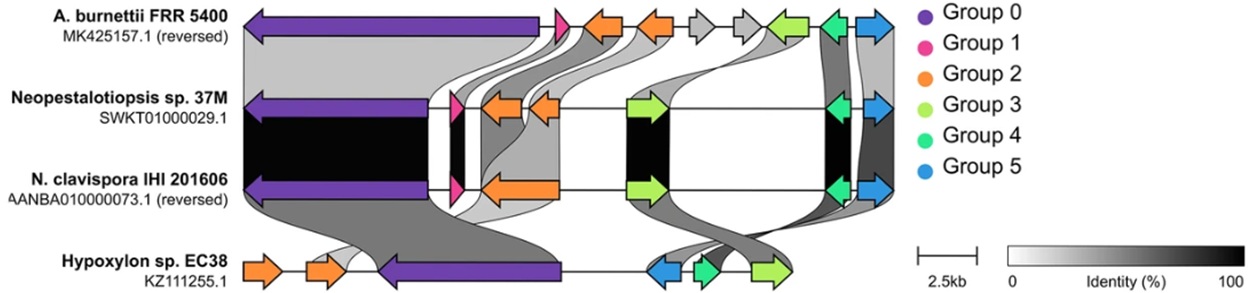
45. Maeda T, Tsuyuki Y, Yoshida H, Goto M, Takahashi T. Characterization of integrative and conjugative elements carrying erm(B) and tet(O) resistance determinants in streptococcus uberis isolates from bovine milk in Chiba prefecture, Japan: CompArative GEne cluster analysis toolbox with ICEfinder. BMC Res Notes 2024;17:377.
46. ICEberg 3.0 project. ICEfinder: Detection of ICE/IME of bacterial genomes. https://tool2-mml.sjtu.edu.cn/ICEberg3/ICEfinder.php [Online] (last visited on 12 September 2025).
47. Lao J, Lacroix T, Guédon G, Coluzzi C, Payot S, Leblond-Bourget N, et al. ICEscreen: a tool to detect Firmicute ICEs and IMEs, isolated or enclosed in composite structures. NAR Genom Bioinform 2022;4:lqac079.
48. Pountain AW, Yanai I. Dissecting microbial communities with single-cell transcriptome analysis. Science 2025;389:eadp6252.


1. Naik S, Kashyap D, Deep J, Darwish S, Cross J, Mansoor E, et al. Utilizing next-generation sequencing: advancements in the diagnosis of fungal infections. Diagnostics (Basel) 2024;14:1664.
2. Li W, Cui Q, Bai L, Fu P, Han H, Liu J, et al. Application of whole-genome sequencing in the national molecular tracing network for foodborne disease surveillance in China. Foodborne Pathog Dis 2021;18:538-46.
3. Gomes E, Araújo D, Nogueira T, Oliveira R, Silva S, Oliveira LVN, et al. Advances in whole genome sequencing for foodborne pathogens: implications for clinical infectious disease surveillance and public health. Front Cell Infect Microbiol 2025;15:1593219.
4. Wesenberg S, Mueller C, Nestmann F, Holthoff-Detto V. Effects of an animal-assisted intervention on social behaviour, emotions, and behavioural and psychological symptoms in nursing home residents with dementia. Psychogeriatrics 2019;19:219-27.
5. Thodberg K, Videbech PB, Hansen TGB, Pedersen AB, Christensen JW. Dog visits in nursing homes – increase complexity or keep it simple? A randomised controlled study. PLoS One 2021;16:e0251571.
6. Centers for Disease Control and Prevention. About One Health (updated on 27 June 2025). https://www.cdc.gov/onehealth/index.html [Online] (last visited on 12 September 2025).
7. Shropshire WC, Hanson BM, Shelburne SA. Genome-wide approaches to bacterial strain typing: a history and review of recent methodological advances. Curr Opin Infect Dis 2025;38:329-38.
8. Maeda T, Yoshida H, Abe N, Murakami K, Goto M, Takahashi T. Draft genome sequence of emm103/ST1363 Streptococcus pyogenes strain AB1, isolated from the blood of a woman with peritonitis and toxic shock syndrome. Microbiol Resour Announc 2024;13:e0102723.
9. Anslan S, Mikryukov V, Armolaitis K, Ankuda J, Lazdina D, Makovskis K, et al. Highly comparable metabarcoding results from MGI-Tech and Illumina sequencing platforms. PeerJ 2021;9:e12254.
10. Yoshida H, Katayama Y, Fukushima Y, Ohtaki H, Ohkusu K, Mizutani T, et al. Draft genome sequence of Streptococcus canis clinical strain OT1, isolated from a dog owner with invasive infection without a dog bite in Japan. Microbiol Resour Announc 2019;8:e00770-19.
11. Fukushima Y, Murata Y, Katayama Y, Tsuyuki Y, Yoshida H, Mizutani T, et al. Draft genome sequence of blood-origin Streptococcus canis strain FU149, isolated from a dog with necrotizing soft tissue infection. Microbiol Resour Announc 2020;9:e00737-20.
12. Kim JS, Sakaguchi S, Fukushima Y, Yoshida H, Nakano T, Takahashi T. Complete genome sequences of four Streptococcus canis strains isolated from dogs in South Korea. Microbiol Resour Announc 2020;9:e00818-20.
13. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34:i884-90.
14. Kolar O, Appleberry H, Wolfe AJ, Kula A, Putonti C. Complete genome sequence of vaginal swab isolate Enterococcus faecalis UMB6935B, including two complete plasmids. Microbiol Resour Announc 2025;14:e0036825.
15. Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A 2005;102:13950-5.
16. He EM, Chen CW, Guo Y, Hsu MH, Zhang L, Chen HL, et al. The genome of serotype VI Streptococcus agalactiae serotype VI and comparative analysis. Gene 2017;597:59-65.
17. EZBioCloud. About Pan-Genome Orthologous Group (POG) (updated on 15 May 2017). https://help.ezbiocloud.net/uncategorized/pan-genome-orthologous-group-pog/ [Online] (last visited on 12 September 2025).
18. Lin G, Chai J, Yuan S, Mai C, Cai L, Murphy RW, et al. VennPainter: a tool for the comparison and identification of candidate genes based on Venn diagrams. PLoS One 2016;11:e0154315.
19. Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016;44:D457-62.
20. Takahashi T, Lee S, Kim S. Genomic characteristics of Streptococcus agalactiae based on the pan-genome orthologous group analysis according to invasiveness and capsular genotype. J Infect Chemother 2021;27:814-9.
21. Kim JM, Fukushima Y, Yoshida H, Kim JS, Takahashi T. Comparative genomic features of Streptococcus canis based on pan-genome orthologous group analysis according to sequence type. Jpn J Infect Dis 2022;75:269-76.
22. Takahashi T, Kim H, Kim HS, Kim HS, Song W, Kim JS. Comparative genomic analysis of staphylococcal cassette chromosome mec type V Staphylococcus aureus strains and estimation of the emergence of SCCmec V clinical isolates in Korea. Ann Lab Med 2024;44:47-55.
23. CompArative GEne Cluster Analysis Toolbox (CAGECAT) project. Welcome to CAGECAT. https://cagecat.bioinformatics.nl [Online] (last visited on 12 September 2025).
24. van den Belt M, Gilchrist C, Booth TJ, Chooi YH, Medema MH, Alanjary M. The CompArative GEne Cluster Analysis Toolbox for rapid search and visualisation of homologous gene clusters. BMC Bioinformatics 2023;24:181.
25. Yoshida H, Kim JM, Maeda T, Goto M, Tsuyuki Y, Shibata S, et al. Virulence-associated genome sequences of Pasteurella canis and unique toxin gene prevalence of P. canis and Pasteurella multocida isolated from humans and companion animals. Ann Lab Med 2023;43:263-72.
26. Virulence Factor Database (VFDB) project. Up-to-date knowledge of VFs of various bacterial pathogens. http://www.mgc.ac.cn/VFs/ [Online] (last visited on 12 September 2025).
27. Hirose Y, Yamaguchi M, Okuzaki D, Motooka D, Hamamoto H, Hanada T, et al. Streptococcus pyogenes transcriptome changes in the inflammatory environment of necrotizing fasciitis. Appl Environ Microbiol 2019;85:e01428-19.
28. Hirose Y, Yamaguchi M, Sumitomo T, Nakata M, Hanada T, Okuzaki D, et al. Streptococcus pyogenes upregulates arginine catabolism to exert its pathogenesis on the skin surface. Cell Rep 2021;34:108924.
29. Yoshida H, Goto M, Tsuyuki Y, Kim JS, Takahashi T. Streptococcus canis transcriptomic modifications in host cell entry environments of human keratinocytes. BMC Genomics 2024;25:1028.
30. Westermann AJ, Gorski SA, Vogel J. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 2012;10:618-30.
31. Deb S, Basu J, Choudhary M. An overview of next generation sequencing strategies and genomics tools used for tuberculosis research. J Appl Microbiol 2024;135:lxae174.
32. Wilkening RV, Langouët-Astrié C, Severn MM, Federle MJ, Horswill AR. Identifying genetic determinants of Streptococcus pyogenes-host interactions in a murine intact skin infection model. Cell Rep 2023;42:113332.
33. Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 1998;95:3140-5.
34. Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 2010;11:595.
35. Uelze L, Grützke J, Borowiak M, Hammerl JA, Juraschek K, Deneke C, et al. Typing methods based on whole genome sequencing data. One Health Outlook 2020;2:3.
36. Silva M, Machado MP, Silva DN, Rossi M, Moran-Gilad J, Santos S, et al. chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. Microb Genom 2018;4:e000166.
37. Seah YM, Stewart MK, Hoogestraat D, Ryder M, Cookson BT, Salipante SJ, et al. In silico evaluation of variant calling methods for bacterial whole-genome sequencing assays. J Clin Microbiol 2023;61:e0184222.
38. Hall MB, Wick RR, Judd LM, Nguyen AN, Steinig EJ, Xie O, et al. Benchmarking reveals superiority of deep learning variant callers on bacterial nanopore sequence data. eLife 2024;13:RP98300.
39. Murray CJL, Ikuta KS, Sharara F, Swetschinski L, Robles Aguilar G, Gray A, et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022;399:629-55.
40. Sherry NL, Lee JYH, Giulieri SG, Connor CH, Horan K, Lacey JA, et al. Genomics for antimicrobial resistance-progress and future directions. Antimicrob Agents Chemother 2025;69:e0108224.
41. Doyle RM, O’Sullivan DM, Aller SD, Bruchmann S, Clark T, Coello Pelegrin A, et al. Discordant bioinformatic predictions of antimicrobial resistance from whole-genome sequencing data of bacterial isolates: an inter-laboratory study. Microb Genom 2020;6:e000335.
42. Burrus V, Pavlovic G, Decaris B, Guédon G. Conjugative transposons: the tip of the iceberg. Mol Microbiol 2002;46:601-10.
43. Huang J, Liang Y, Guo D, Shang K, Ge L, Kashif J, et al. Comparative genomic analysis of the ICESa2603 family ICEs and spread of erm(B)- and tet(O)-carrying transferable 89K-subtype ICEs in swine and bovine isolates in China. Front Microbiol 2016;7:55.
44. Beres SB, Musser JM. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One 2007;2:e800.
45. Maeda T, Tsuyuki Y, Yoshida H, Goto M, Takahashi T. Characterization of integrative and conjugative elements carrying erm(B) and tet(O) resistance determinants in streptococcus uberis isolates from bovine milk in Chiba prefecture, Japan: CompArative GEne cluster analysis toolbox with ICEfinder. BMC Res Notes 2024;17:377.
46. ICEberg 3.0 project. ICEfinder: Detection of ICE/IME of bacterial genomes. https://tool2-mml.sjtu.edu.cn/ICEberg3/ICEfinder.php [Online] (last visited on 12 September 2025).
47. Lao J, Lacroix T, Guédon G, Coluzzi C, Payot S, Leblond-Bourget N, et al. ICEscreen: a tool to detect Firmicute ICEs and IMEs, isolated or enclosed in composite structures. NAR Genom Bioinform 2022;4:lqac079.
48. Pountain AW, Yanai I. Dissecting microbial communities with single-cell transcriptome analysis. Science 2025;389:eadp6252.